-

Autoimmunität bei membranöser Nephropathie
Krankheitsbild und Ursachen der membranösen Nephropathie
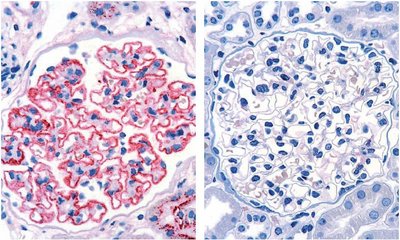
Die membranöse Nephropathie (MN, auch: membranöse Glomerulonephritis) ist eine chronisch entzündliche Erkrankung der Nierenkörperchen (Glomeruli), die durch eine Verdickung der glomerulären Kapillarwände aufgrund von Immunkomplexablagerungen gekennzeichnet ist (siehe Abbildung). Durch die Ablagerungen kommt es zur Schädigung der Podocyten und Störung der Permeabilität der glomerulären Basalmembran, was zu einer Proteinurie führt. Steigt die Proteinausscheidung im Urin stark an (>3.5 g/24 h), kann ein nephrotisches Syndrom mit verminderter Proteinkonzentration im Blut (Hypoproteinämie), erhöhten Blutfettwerten (Hyperlipidämie) und Ödemen entstehen.1
 |
Ungefähr 20-30 % der MN-Fälle entstehen sekundär als Folge verschiedener Grunderkrankungen oder der Anwendung bestimmter Medikamente.1,2 Sie müssen von der primären MN abgegrenzt werden, bei der Autoantikörper zur Entstehung der Krankheit führen (siehe Tabelle). PLA2R und THSD7A sind die ersten Antigene, die als Zielstrukturen solcher Autoantikörper entdeckt wurden. Liegen weder andere ursächliche Erkrankungen noch detektierbare Autoantikörper vor, wird von idiopathischen MN-Fällen gesprochen.3
| Ursachen für pMN |
|---|
| Autoantikörper (PLA2R/THSD7A) |
| Ursachen für sMN4 |
|---|
| Infektionen (z. B. Hepatitis B Virus, Hepatitis C Virus, Syphilis, Malaria) |
| Autoimmunerkrankungen (z. B. Rheumatoide Arthritis, Sjögren´s Syndrom, Bullöses Pemphigoid) |
| Krebserkrankungen |
| Sonstige Erkrankungen (z. B. Sichelzellanämie, Guillain-Barré-Syndrom) |
| Medikamente (z. B. nichtsteroidale Entzündungshemmer (NSAID), Gold, Quecksilber, Penicillamin) |
FAZIT: Da die Therapie der beiden Krankheitsformen sehr unterschiedlich erfolgt, ist die diagnostische Unterscheidung zwischen der primären und sekundären Form der MN von großer klinischer Bedeutung.2 Während sich bei der sMN die Therapie auf die zugrunde liegende Erkrankung fokussiert, wird bei der Behandlung von Patienten mit pMN hauptsächlich eine immunsupprimierende Strategie angewendet. Daher kann eine korrekte und schnelle Diagnose unnötige diagnostische Prozeduren oder Medikamenteneinnahmen vermeiden.
Entdeckung und pathogene Rolle der Autoantikörper
2009 identifizierte die Arbeitsgruppe um David J. Salant PLA2R, ein 185-kDa Glykoprotein, als spezifisches Autoantigen für Antikörper, die in 70-80 % aller Patienten mit MN ohne sekundäre Ursache nachgewiesen werden konnten.3 2014 wurde mit THSD7A ein zweites Antigen in Zusammenhang mit primärer MN beschrieben.5 Die Prävalenz von Antikörpern gegen THSD7A wird mit Werten von bis zu 10 % angegeben. Auch wenn Antikörper gegen PLA2R und THSD7A in seltenen Fällen parallel auftreten können, so wurden Anti-THSD7A-Antikörper vorwiegend in anti-PLA2R-seronegativen pMN-Patienten gefunden.6 In Patienten mit sMN konnten weder PLA2R- noch THSD7A-Autoantikörper nachgewiesen werden.
Die Antigene werden auf der Oberfläche der Podozyten exprimiert. Sie binden zirkulierende Autoantikörper und es bilden sich Immunkomplex-Ablagerungen.
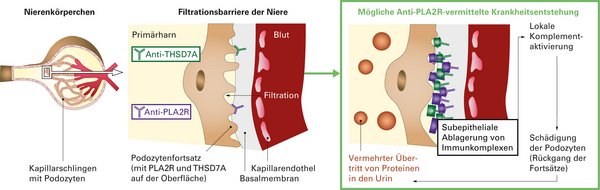 |
Der pathogene Mechanismus der Entstehung der pMN ist bisher noch nicht vollständig geklärt. Konsens besteht, dass die Verdickung der glomerulären Kapillarwände durch die abgelagerten Immunkomplexe verursacht wird. Diese Ablagerungen lösen lokale Komplementaktivierungen aus, welche über die Involvierung verschiedener Mediatoren zu einer Degradation des Zytoskeletts der Podocyten führt. Die gestörten Podocyten verlieren ihre Filtrationsfunktion und sezernieren darüber hinaus zusätzliche Faktoren, die sich negativ auf die glomeruläre Basalmembran auswirken.1 Es kommt zur fortschreitenden Störung der Filterfunktion und somit zu einem vermehrten Übertritt von Eiweiß in den Primärharn (Proteinurie) sowie zur Entstehung von Ödemen und einer kompensatorischen Steigerung der Lipoproteinsynthese, was in einem nephrotischen Syndrom resultieren kann.
Die Relevanz der Autoantikörper an der Pathogenese der pMN konnte 2017 für THSD7A im Mausmodell gezeigt werden. Durch die Injektion von THSD7A-gerichteter Antikörper wurde ein schweres nephrotisches Syndrom mit Proteinurie und Hyperlipidämie in Mäusen ausgelöst.7
Während die durch die Autoantikörper ausgelöste Pathogenese mechanistisch gleich ist, wurden in der Klinik von anti-PLA2R- und anti-THSD7A-positiven pMN-Patienten Unterschiede beobachtet. Eine Untersuchung von Hoxha et al. (2016) zeigte ein gehäuftes Auftreten bösartiger Tumore in anti-THSD7A-positiven Patienten.8
1Ronco et al., Nat Rev Dis Primers 7, 69 (2021)
2KDIGO, Kidney inter. 100 (Suppl.): 1–276 (2021)
3Beck et al., N Engl J Med 361, 11-21 (2009)
4Trujillo et al., Nephron. 144, 261-271 (2020)
5Tomas et al., N Engl J Med 371, 2277-2287 (2014)
6Tomas et al., J Am Soc Nephrol 28, 3262-3277 (2017)
7Larsen et al., Mod Pathol 29, 421-426 (2016)
8Hoxha et al., J Am Soc Nephrol 28, 520-531 (2017)
Ärzte & Labore
Informationsmaterial
 Diagnosestellung ohne Biopsie - Jetzt Video anschauen!
Diagnosestellung ohne Biopsie - Jetzt Video anschauen!